지텔만 증후군
"오늘의AI위키"의 AI를 통해 더욱 풍부하고 폭넓은 지식 경험을 누리세요.
1. 개요
지텔만 증후군은 SLC12A3 유전자의 돌연변이로 인해 발생하는 드문 유전 질환으로, 신장의 원위세뇨관에서 전해질 재흡수에 이상이 생겨 저칼륨혈증, 저마그네슘혈증, 대사성 알칼리증 등의 증상을 보인다. 이 질환은 상염색체 열성 유전 방식으로 유전되며, 유병률은 인구 100만 명당 약 25명으로 추정된다. 증상은 근육 경련, 쇠약, 갈증, 야뇨증 등 다양하게 나타나며, 심한 경우 심장 부정맥을 유발할 수 있다. 진단은 혈액 및 소변 검사를 통해 이루어지며, 바터 증후군 등 다른 질환과의 감별이 중요하다. 치료는 마그네슘 및 칼륨 보충, 고염분 식단 조절, 필요에 따라 칼륨 보존성 이뇨제 투여 등을 통해 증상을 완화하는 방향으로 진행된다.
더 읽어볼만한 페이지
- 막 수송 단백질 장애 - 크론병
크론병은 위장관에 만성 염증을 일으키는 질환으로, 유전적 요인, 면역계 이상, 환경적 요인이 복합적으로 작용하며 복통, 설사, 합병증 등을 유발할 수 있고, 약물 요법, 영양 요법, 수술 등으로 치료하며 완치 치료법은 없다. - 막 수송 단백질 장애 - 부신백질이영양증
부신백질이영양증은 X 염색체 *ABCD1* 유전자 돌연변이로 초장쇄 지방산 수송 장애가 발생, 뇌 백질, 부신 피질 등에 초장쇄 지방산이 축적되어 신경학적 증상과 부신 기능 부전을 유발하는 유전 질환이다. - 신장 비뇨기 질환 - 신장염
신장염은 콩팥에 발생한 염증으로, 자가면역질환, 요로감염, 루푸스, 격렬한 운동 등이 원인이 되어 혈뇨, 핍뇨, 요독증, 고혈압 등의 증상을 유발하며, 혈액 검사, X레이, 초음파 등으로 진단하고 원인에 따라 치료한다. - 신장 비뇨기 질환 - 신우신염
신우신염은 신장의 신우와 신실질에 발생하는 감염으로, 급성(고열, 배뇨통, 옆구리 통증)과 만성(지속적인 옆구리 통증, 체중 감소)으로 나뉘며, 대장균과 장구균이 주요 원인균이고, 항생제로 치료하며, 요로의 구조적 이상, 당뇨병, 면역 저하 등이 위험 요인이다. - 상염색체 열성 질환 - 윌슨병
윌슨병은 구리가 간과 뇌에 축적되어 발생하는 유전 질환으로, ATP7B 유전자 돌연변이가 원인이며, 간 기능 장애, 신경정신과적 증상, 카이저-플라이셔 고리 등의 증상이 나타난다. - 상염색체 열성 질환 - 색소성 건피증
색소성 건피증은 자외선에 손상된 DNA 복구 기능 저하로 발생하는 유전 질환으로, 햇빛 과민반응, 피부암, 신경계 이상을 유발하며, 자외선 차단과 피부암 예방을 주요 치료 목표로 하는 '어둠의 아이들'이라고도 불리는 질환이다.
| 지텔만 증후군 | |
|---|---|
| 일반 사항 | |
| 동의어 | 일차성 신세뇨관 저칼륨혈증 저마그네슘혈증 저칼슘뇨증 |
| 분야 | 신장학 |
| 원인 | SLC12A3 유전자 돌연변이 CLCKNB 유전자 돌연변이 MT-TI 유전자 돌연변이 MT-TF 유전자 돌연변이 |
 | |
| 추가 정보 | |
| 발견 | 1966년 길텔만 등이 보고 |
2. 발병
대부분의 지텔만 증후군 환자들에서 ''SLC12A3'' 유전자의 돌연변이가 관찰된다. 이 돌연변이는 ''SLC12A3'' 유전자가 티아지드계 이뇨제에 작용하는(thiazide-sensitive) NaCl 공동수송체(cotransporter, 이하 NCC)를 부호화하는 기능을 상실하게 만든다. NCC는 네프론(신단위)의 한 부분인 원위세뇨관에서의 전해질 항상성을 조절하는 역할을 한다.
지텔만 증후군 환자의 증상은 장기적인 티아지드계 이뇨제 처방을 받는 환자들의 증상과 유사하며,[33] 저마그네슘혈증, 저칼슘뇨증, 속발성 알도스테론증에 의한 저칼륨혈증, 대사성 알칼리증이 동반되는 것이 특징이다.[33]
지텔만 증후군의 진단은 저칼륨혈증과 대사성 알칼리증의 다른 흔한 병리학적 원인을 배제한 후에 확진될 수 있다.[19] 전해질 수치를 평가하기 위해 완전 대사 패널(CMP) 또는 기본 대사 패널(BMP)을 사용할 수 있다. 소변을 통해 전해질 측정 및 알도스테론 수치를 측정할 수 있다.[19] 특징적인 임상 지표로는 소변 배설의 결과로 혈액 내 칼륨, 나트륨, 염소 및 마그네슘의 낮은 혈청 수치가 있다.[18] 소변 분획 배설 칼륨은 저칼륨혈증의 맥락에서 높거나 부적절하게 정상이며, 높은 수준의 소변 나트륨과 염소가 관찰된다. 다른 임상 지표로는 혈류 내 혈청 레닌 및 알도스테론 증가와 대사성 알칼리증이 있다. 이 증후군의 증상적 특징은 무증상에서 경미한 증상(쇠약, 경련)에서 심각한 증상(강직, 마비, 횡문근융해증)까지 매우 다양하다.[19] 증상 심각도는 다인자적이며, 표현형 발현은 동일 가족 내의 개인마다 다르다. 유전자 검사는 질병의 병리학적 증상을 유발하는 근본적인 돌연변이를 식별하는 또 다른 방법이다. 이 검사 방식은 일부 실험실에서 이용 가능하다.[19]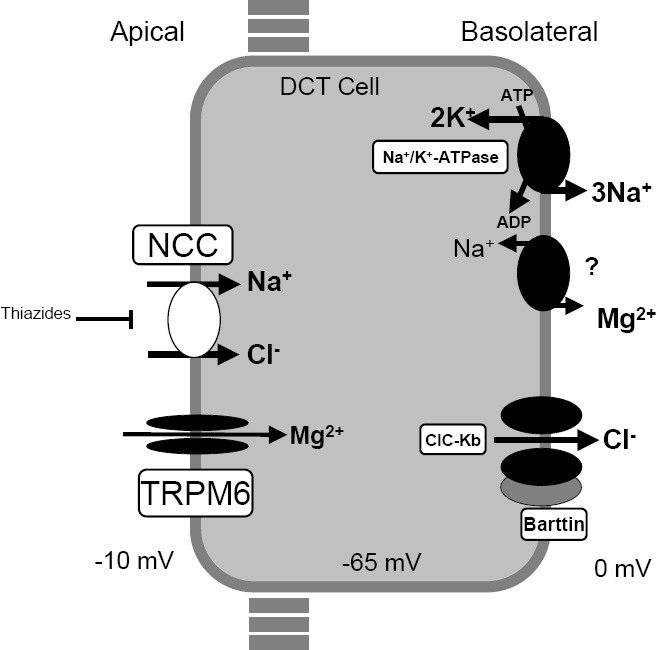
정상인의 신장 내의 원위세뇨관에서는 염화나트륨(NaCl)이 NCC를 통해 세포로 들어오고, 기저측면 Cl- 이온 채널(basolateral Cl- channel (ClC-Kb))과 Na+/K+ ATP효소(ATPase)를 통해 세포를 빠져나가야 한다. 그리고 이러한 과정은 꼭지면 막(apical membrane)에서 발견되는 TRPM6 Mg2+ 이온 채널과 기저측면 막(basolateral membrane)에 있는 추정상 Na/Mg 이온 교환체(putative Na/Mg exchanger)에 의해 조절되어야 한다. 그러나 지텔만 증후군에 걸린 환자는 NCC의 기능 소실로 인해 이러한 과정이 원활하지 않다.
따라서 지텔만 증후군 환자는 전해질 항상성이 유지되지 않으며, 각종 전해질 이상 질환이 수반된다. 지텔만 증후군은 상염색체 열성 유전 질환으로 결함이 있는 대립유전자가 부모 유전자에 각각 존재할 때 발병하게 된다.
유병율은 100만명 당 25명으로 추정되고, 백인 인구의 약 1%가 이형접합체를 가지고 있다.
3. 증상
대사성 알칼리증, 저칼륨혈증이 주요 증상이며,[33] 저칼슘뇨증과 저마그네슘혈증은 대부분의 환자에게서 나타나지만 일부 환자에서는 나타나지 않을 수 있다.[33][2] 고든 증후군 환자와 달리, 지텔만 증후군 환자는 정상 혈압 또는 약간의 저혈압을 보인다.[33][2]
환자들은 심한 근육 경련이나 쇠약, 무감각, 갈증, 야뇨증, 염분 갈망, 이상감각, 연골석회화증, 극심한 피로 또는 과민성으로 나타나는 쇠약을 호소하며,[2] 염분에 대한 갈망이 가장 흔하고 심하다.[6] 발작, 테타니, 마비와 같은 더 심각한 증상도 보고되었다.[2] 저칼륨혈증과 저마그네슘혈증은 심장 근육세포의 활동전위를 연장시켜 비정상적인 심장 리듬과 연장된 QT 간격을 유발하고, 심실성 부정맥의 발생 위험을 높일 수 있다.[33][2] 낮은 칼륨 수치로 인한 급성 심장사 사례도 보고되었다.[2]
환자 간 증상 차이는 유전적 환경의 차이와 NCC 단백질 변이 과정에서의 특정 아미노산에 따른 결과로 보인다.[34] 심각한 표현형 발현을 보이는 지텔만 증후군 환자의 하위 집단에서는 신경근 증상, 성장 지연 및 심실성 부정맥이 관찰되었다.[8] 저마그네슘혈증은 졸음, 쇠약감, 메스꺼움, 구토, 인격 변화를 일으킬 수 있다.
4. 원인
지텔만 증후군은 대부분 나트륨-염소 공동수송체(NCC)를 암호화하는 ''SLC12A3'' 유전자의 돌연변이로 인해 발생한다.[9] 이 돌연변이는 NCC의 기능 상실을 초래하여, 네프론(신단위)의 한 부분인 원위세뇨관에서의 전해질 항상성 조절 기능을 저하시킨다.[2] 정상적인 원위세뇨관에서는 염화나트륨(NaCl)이 NCC를 통해 세포 내로 유입된 후, 기저측면 Cl- 이온 채널(ClC-Kb)과 Na+/K+ ATP효소(ATPase)를 통해 세포 밖으로 배출된다. 이 과정은 꼭지면 막의 TRPM6 Mg2+ 이온 채널과 기저측면 막의 Na/Mg 이온 교환체에 의해 조절된다. 그러나 지텔만 증후군 환자는 NCC 기능 소실로 인해 이러한 과정이 원활하게 진행되지 않아 전해질 불균형이 발생한다.
''SLC12A3'' 유전자에는 미스센스 변이, 넌센스 변이, 프레임 시프트 변이, 스플라이스 부위 변이, 인트론 변이 등 다양한 유형의 돌연변이가 보고되었으며,[10][11] 2012년에는 180개 이상의 돌연변이가 보고되었다.[2]
NCC 기능 상실은 원위 세뇨관에서 나트륨 및 염소 재흡수를 감소시켜 혈압을 낮출 수 있다.[12] 또한, 원위 세뇨관의 길이 단축,[13] 세포간 칼슘 재흡수 증가,[15] 2가 양이온 재흡수 변화[14] 등을 유발할 수 있다.
비활성화된 NCC는 레닌-안지오텐신 알도스테론 시스템(RAAS)을 활성화시킨다.[19] RAAS 활성화는 원위 세뇨관에서 나트륨과 염소 재흡수 실패로 인한 세포 탈수를 보상하기 위한 반응으로, 혈청 칼륨 수치를 낮추는 결과를 초래한다.
일부 지텔만 증후군 환자는 ''SLC12A3'' 유전자에서 유전적 결함이 발견되지 않는데, 이 경우 다른 유전적 결함이 원인일 수 있으며, 일부는 특발성으로 남아있다.[16][17] 지텔만 증후군은 상염색체 열성 유전 질환으로, 결함이 있는 대립유전자가 부모 양쪽 모두에게서 유전될 때 발병한다.
5. 진단
일반적인 진단을 통해 하나의 병원성 변이체만 발견될 경우, ''SLC12A3'' 인트론을 선별하는 것을 고려할 수 있다.[11]
전해질 이상에 대한 감별 진단을 배제하기 위한 검사가 중요하다.[20][21]
5. 1. 감별 진단
지텔만 증후군은 다음과 같은 질병들과 감별해야 한다.[22]
6. 치료
지텔만 증후군은 유전적 결함에 의한 질환이므로 근본적인 치료는 불가능하며, 증상 완화를 위한 대증 요법이 주된 치료 방법이다.[35] 저칼륨혈증, 저마그네슘혈증을 억제하기 위해 산화 마그네슘(MgO)과 같은 마그네슘 제재와 염화 칼륨(KCl) 등의 칼륨 제재를 꾸준히 섭취하고, 고나트륨 및 고칼륨 식이를 하는 것이 일반적이다.[35] 이러한 치료를 통해 환자는 주기성 사지마비나 부정맥 등의 심각한 증상을 예방할 수 있다.[35]
칼륨 제재로 저칼륨혈증이 교정되지 않을 경우, 칼륨 보존성 이뇨제(아밀로라이드 등) 및 알도스테론 억제제 투여를 고려할 수 있다.[36] 알도스테론 길항제(예: 스피로노락톤 또는 에플레레논) 또는 상피 나트륨 채널 차단제(예: 아밀로라이드)도 칼륨의 요로 손실을 감소시키기 때문에 가능한 치료법으로 제시되었다. 그러나 2017년 전문가 합의 성명에서는 이러한 약물이 부작용의 가능성 때문에 지텔만 증후군 환자에게 주의해서 사용해야 한다고 경고했다.[21]
전해질 불균형과 관련된 증상 완화를 위해 칼륨과 마그네슘 보충과 함께 고염분 식단 조절이 필요하다.[2] 소변으로 손실되는 전해질을 적절히 대체하기 위해 칼륨과 마그네슘을 고용량으로 투여해야 하는 경우가 많다.[2] 경구 마그네슘의 흔한 부작용인 설사를 줄이기 위해 하루 3~4회로 나누어 복용하는 것이 좋다.[2] 심각한 전해질 불균형의 경우 정맥 내 투여가 필요할 수 있다.[2]
성장 지연이 있는 환자의 경우 성장 호르몬 치료를 병행하면 성장 지연과 저마그네슘혈증 개선에 도움이 될 수 있다는 보고가 있다.[36] 영아 및 소아와 같이 질병의 발병이 빠른 환자의 경우 성장 장애를 치료하기 위해 인도메타신이 사용되기도 한다. 그러나 인도메타신은 사구체 여과율 감소 및 위장 장애와 같은 부작용이 있을 수 있어 주의해서 사용해야 한다.[21][24]
부정맥 예방과 QT 간격 활동 모니터링을 위한 심장 평가가 권장된다.[19] QT 간격을 연장하거나 지연시키는 약물(마크로라이드, 항히스타민제, 베타-2 작용제)은 심장 사망을 예방하기 위해 피해야 한다.[3]
7. 역학
지텔만 증후군의 유병률은 인구에 따라 8만 명 중 1명에서 500명 중 1명까지 다양하게 추정된다.[25][26] 남녀 발병 비율은 1:1이다. 이 질환은 일반적으로 10대 이후, 즉 청소년기 또는 성인기에 발생하지만, 신생아기에도 발생할 수 있다. ''SLC12A3'' 유전자 변이의 이형 접합 보인자는 인구의 1% 정도이다.[19] 지텔만 증후군 환자가 자녀에게 질병을 물려줄 확률은 낮으며, 부모가 모두 보인자가 아닌 경우 약 400분의 1이다.[9]
8. 역사
지텔만 증후군은 신장학 전문의 힐렐 조나단 지텔만(Hillel Jonathan Gitelman영어, 1932년~2015년 1월 12일)의 이름을 따서 명명되었으며, 그는 노스캐롤라이나 대학교 의과대학에서 근무했다.[27][28] 지텔만은 1966년 이 질환을 가진 자매를 관찰한 후 처음으로 이 질환을 설명했다.[29] 지텔만과 그의 동료들은 이후 분자 클로닝을 통해 원인이 되는 유전자(''SLC12A3'')를 확인하고 분리했다.[30]
참조
[1]
Citation
English: This is an image of a kidney nephron and its structure.
https://commons.wiki[...]
2013-01-31
[2]
Review
Gitelman's syndrome: a pathophysiological and clinical update
2012-02
[3]
학술지
Bartter- and Gitelman-like syndromes: salt-losing tubulopathies with loop or DCT defects
2011-10
[4]
학술지
Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia
2005-06
[5]
학술지
Salt handling and hypertension
2004-04
[6]
학술지
Gitelman syndrome in a South African family presenting with hypokalaemia and unusual food cravings
2017-01
[7]
학술지
Gitelman's syndrome revisited: an evaluation of symptoms and health-related quality of life
2001-02
[8]
학술지
Transcriptional and functional analyses of SLC12A3 mutations: new clues for the pathogenesis of Gitelman syndrome
2007-04
[9]
학술지
Gitelman syndrome
BioMed Central Ltd
2008-07
[10]
학술지
Spectrum of mutations in Gitelman syndrome
2011-04
[11]
학술지
Long-Read Sequencing Identifies Novel Pathogenic Intronic Variants in Gitelman Syndrome
2023-02
[12]
학술지
Mutations in the Na-Cl cotransporter reduce blood pressure in humans
2001-06
[13]
학술지
Altered renal distal tubule structure and renal Na(+) and Ca(2+) handling in a mouse model for Gitelman's syndrome
2004-09
[14]
학술지
Mechanisms coupling sodium and magnesium reabsorption in the distal convoluted tubule of the kidney
2021-02
[15]
학술지
The mechanism of hypocalciuria with NaCl cotransporter inhibition
2011-09
[16]
학술지
Phenotypic differences of mutation-negative cases in Gitelman syndrome clinically diagnosed in adulthood
2021-03
[17]
학술지
The genetic spectrum of Gitelman(-like) syndromes
2022-09
[18]
학술지
Renal phosphate handling in Gitelman syndrome--the results of a case-control study
https://boris.unibe.[...]
2013-01
[19]
웹사이트
Gitelman Syndrome
https://rarediseases[...]
2020-03-29
[20]
학술지
The challenges of diagnosis and management of Gitelman syndrome
2020-01
[21]
학술지
Gitelman syndrome: consensus and guidance from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference
2017-01
[22]
학술지
Genetic causes of hypomagnesemia, a clinical overview
2017-07
[23]
학술지
Gitelman-Like Syndrome Caused by Pathogenic Variants in mtDNA
2022-02
[24]
학술지
Indomethacin, amiloride, or eplerenone for treating hypokalemia in Gitelman syndrome
2015-02
[25]
학술지
Resistance to Insulin in Patients with Gitelman Syndrome and a Subtle Intermediate Phenotype in Heterozygous Carriers: A Cross-Sectional Study
2019-08
[26]
학술지
Examination of the predicted prevalence of Gitelman syndrome by ethnicity based on genome databases
2021-08
[27]
명명
synd/2329
[28]
웹사이트
Hillel J. Gitelman '54
https://paw.princeto[...]
Princeton Alumni Weekly
2015-05-13
[29]
학술지
A new familial disorder characterized by hypokalemia and hypomagnesemia
[30]
학술지
Bartter's and Gitelman's syndromes: their relationship to the actions of loop and thiazide diuretics
http://147.83.15.91/[...]
2006-04
[31]
WhoNamedIt
synd/2329
http://www.whonamedi[...]
[32]
저널 인용
A new familial disorder characterized by hypokalemia and hypomagnesemia
[33]
저널 인용
Salt handling and hypertension
[34]
저널 인용
Bartter's and Gitelman's syndromes: from gene to clinic
[35]
URL
http://www.snubi.org[...]
[36]
웹인용
보관된 사본
https://web.archive.[...]
2014-11-17
본 사이트는 AI가 위키백과와 뉴스 기사,정부 간행물,학술 논문등을 바탕으로 정보를 가공하여 제공하는 백과사전형 서비스입니다.
모든 문서는 AI에 의해 자동 생성되며, CC BY-SA 4.0 라이선스에 따라 이용할 수 있습니다.
하지만, 위키백과나 뉴스 기사 자체에 오류, 부정확한 정보, 또는 가짜 뉴스가 포함될 수 있으며, AI는 이러한 내용을 완벽하게 걸러내지 못할 수 있습니다.
따라서 제공되는 정보에 일부 오류나 편향이 있을 수 있으므로, 중요한 정보는 반드시 다른 출처를 통해 교차 검증하시기 바랍니다.
문의하기 : help@durumis.com